|

- Home
- BioZoom
- BioZoom archives
- 2016
- 2016 Issue 4
- Precision epigenome editing - promise and challenge
Precision epigenome editing - promise and challenge |
Author: Lasse Fløe, MSc student, and Yonglun Luo, associate professor, Department of Biomedicine, Aarhus University, Bartholins Allé 4, 8000, Aarhus C, laflo@biomed.au.dk, alun@biomed.au.dk.
|
Epigenetic alterations are associated with the pathogenesis of many human diseases such as cancers, diabetes, and neurodegenerative diseases. Chemical treatments have been applied to modify the epigenome as ways of diminishing disease symptoms. However, these approaches are unspecific and cause various unwanted side effects. Recent developments of artificial, programmable epigenetic modifiers based the on CRISPR-associated proteins provide new hopes for efficient and precise control, modification of the epigenome, and potentially human disease therapy. The human epigenome is a highly organized and complex system composed of hierarchical modifications of DNA and histones. These modifications are known as epigenetic marks. Tight temporal and spatial control of these epigenetic marks ensure the proper gene expression and cellular functions.Epigenetic modifications include DNA methylation of cytosines predominantly in CpG dinucleotides, acetylation or methylation of the histone tails, and nucleosome positioning. The modifications play a decisive role in the regulation of gene expression and are linked to the pathogenesis of many human diseases (1). The molecular mechanisms involved in the dynamic regulation of epigenetic modifications are complicated and are associated with many regulatory proteins and enzymes. However, there are still some so-called sole catalytic enzymes carrying out the key-processing steps of epigenetic modifications. For example, de novo DNA methylation is controlled by the DNA methyltransferases (e.g. DNMT3A, DNMT3B, DNMT3L). The ten-eleven translocation (TET) family of proteins such as TET1 is essential for the initial activation of methylated cytosine (5mC) demethylation. Histone acetyltransferases (HAT) such as P300/CBP family enzymes can acetylate lysine at the histone tails. Histone methylation is catalyzed by a class of enzymes called histone methyltransferase (HMT), which transfer one to three methyl groups to lysine or arginine residues of histone proteins. Epigenetic-modifying drugs are available, but unspecific The fundamental correlation between epigenetic modifications and their consequences on gene expression and/or the pathogenesis of human diseases are based on association studies or the application of epigenetic modifying drugs such DNA methyltransferase inhibitor 5-azacytidine and histone deacetylase inhibitor valproic acid. Several of these epigenetic-modifying drugs are making their way into therapeutics. Valproic acid has been used to treat epilepsy, migraines and bipolar disease for many years. But there are new epigenetic-modifying drugs in the pipeline as well such as Chidamide and Belinostat which are being approved for treatment of peripheral T-cell lymphoma. However, most of these drugs generally cause a global epigenome remodeling. For example, treatment of cells with valproic acid results in global histone acetylation, which might have some unprecedented effects on cellular functions. To investigate the biological consequences resulting from specific epigenetic modifications at a certain genomic locus, great efforts have been spent during the last decade to develop molecular tools that can be programmed to specifically modify user-designed genomic loci, also known as “precision epigenome editing tools”. Zink-finger proteins, TALE proteins and the CRISPR-Cas9 are precise epigenome editing tools To date, there are three classes of such tools available in the precision epigenome editing toolbox. All these tools share one fundamental molecular principle: They are made by fusing epigenome-modifying enzymatic proteins or their catalytic domains to programmable DNA binding proteins. These programmable DNA binding proteins include the zinc-finger proteins (ZFs), the transcriptional activator-like effector proteins (TALE), and the nuclease deficient clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (dCas9). The newly developed CRISPR-Cas9 system was originally discovered as an innate immune system in bacteria and archaea. Through molecular engineering, the bacterial CRISPR-Cas9 system has been successfully harnessed for genome editing in almost all kinds of cells and organisms (2). As illustrated in Figure 1, both ZF- and TALE-based epigenome editing tools are based on the dogma of specific protein-DNA recognition, whereas dCas9-based epigenome editing tools include complimentary base pairing between a small, programmable guide RNA (gRNA) and the DNA. Thus, the same dCas9-based fusion proteins with epigenome editing characteristics can be programmed to almost any genomic locus simply by using another gRNA. On the contrary, the ZF- and TALE-based epigenome editing fusion proteins have to be re-engineered for each specific locus, which is time-consuming, laborious and expensive. This makes harnessing the CRISPR-dCas9 system for epigenome editing the most promising and attractive tool for studying epigenetics, as well as providing a potential strategy for targeted epigenome therapy.
A promising tool for efficient and precise editing Precision epigenome editing by CRISPR-dCas9 has been proven promising. This technology provides an alternative, robust approach to modulate gene expression and cell fate. Several studies have shown that targeted epigenetic modifications of specific genomic loci can be achieved through the RNA-guided dCas9-based artificial epigenetic enzymes. For example, dCas9 fused to the N-terminus of the catalytic core of the human acetyltransferase p300, the tetrameric VP16 transcription activator domain (VP64), the krüppel-associated box repressor domain (KRAB), the histone demethylase LSD1, or the DNA methyltransferase domain of DNMT3A have been used for either targeted histone acetylation, histone methylation or DNA methylations (3-5). All these studies have consistently shown that programmable and targeted epigenetic modifications can be achieved in cells through co-delivery of dCas9-based artificial enzymes and gRNAs. Subsequent effects on alteration of gene expression and cell fate have been achieved, if a critical epigenetic regulatory region is modified. Still a problem with off-target recognition However, the applications of CRISPR-dCas9-based precision epigenome editing tools are still facing one critical challenge: Off-targets. Off-targets can result from a combination of dCas9-dependent, gRNA-dependent, or catalytic enzyme-dependent factors. Epigenome editing by dCas9-based epigenetic-modifying enzymes is simply based on the binding of dCas9 to a specific DNA locus. One unique feature of dCas9 is that this protein can rapidly interrogate with DNA through interaction with a protospacer adjacent motif (PAM) across the genome. The PAM sequences are unique for each Cas9 ortholog, e.g. 5’-NGG for the Cas9 protein from Streptococcus pyogenes. Thus, if a highly active catalytic domain was fused to dCas9, the transient interaction between dCas9 and DNA might create substantial un-intended modifications of the epigenome. Furthermore, the catalytic domains of epigenetic modifying enzymes per se could also unspecifically modify the epigenome, simply due to random protein-DNA interaction. Studies based on chromatin immunoprecipitation (ChIP) with massive parallel DNA sequencing (ChIP-seq) have revealed that dCas9 can still strongly bind to DNA even with over 10 mismatches between gRNA guide sequences and the off-target locus. Consistent with this, we have observed all aforementioned off-target effects in an on-going project in which we harness the dCas9-DNMT3A fusion protein for targeted inhibition of oncogenes in cancer cells. The ultimate aim of these epigenetic tools is not merely for perturbation of gene functions but for precision epigenetic therapy of human diseases such as cancers. A better understanding of the cause of these off-target effects might enable us to further improve epigenome editing tools with higher specificity and efficacy. References 1. Portela A and M Esteller. Epigenetic modifications and human disease. Nat Biotechnol 2010;28:1057-68. 2. Sander JD and JK Joung. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 2014;32:347-55. 3. Kearns NA, H Pham, B Tabak, RM Genga, NJ Silverstein, M Garber and R Maehr. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 2015;12:401-3. 4. Hilton IB, AM D’Ippolito, CM Vockley, PI Thakore, GE Crawford, TE Reddy and CA Gersbach. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 2015;33:510-7. 5. Vojta A, P Dobrinic, V Tadic, L Bockor, P Korac, B Julg, M Klasic and V Zoldos. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44(12):5615-28 |
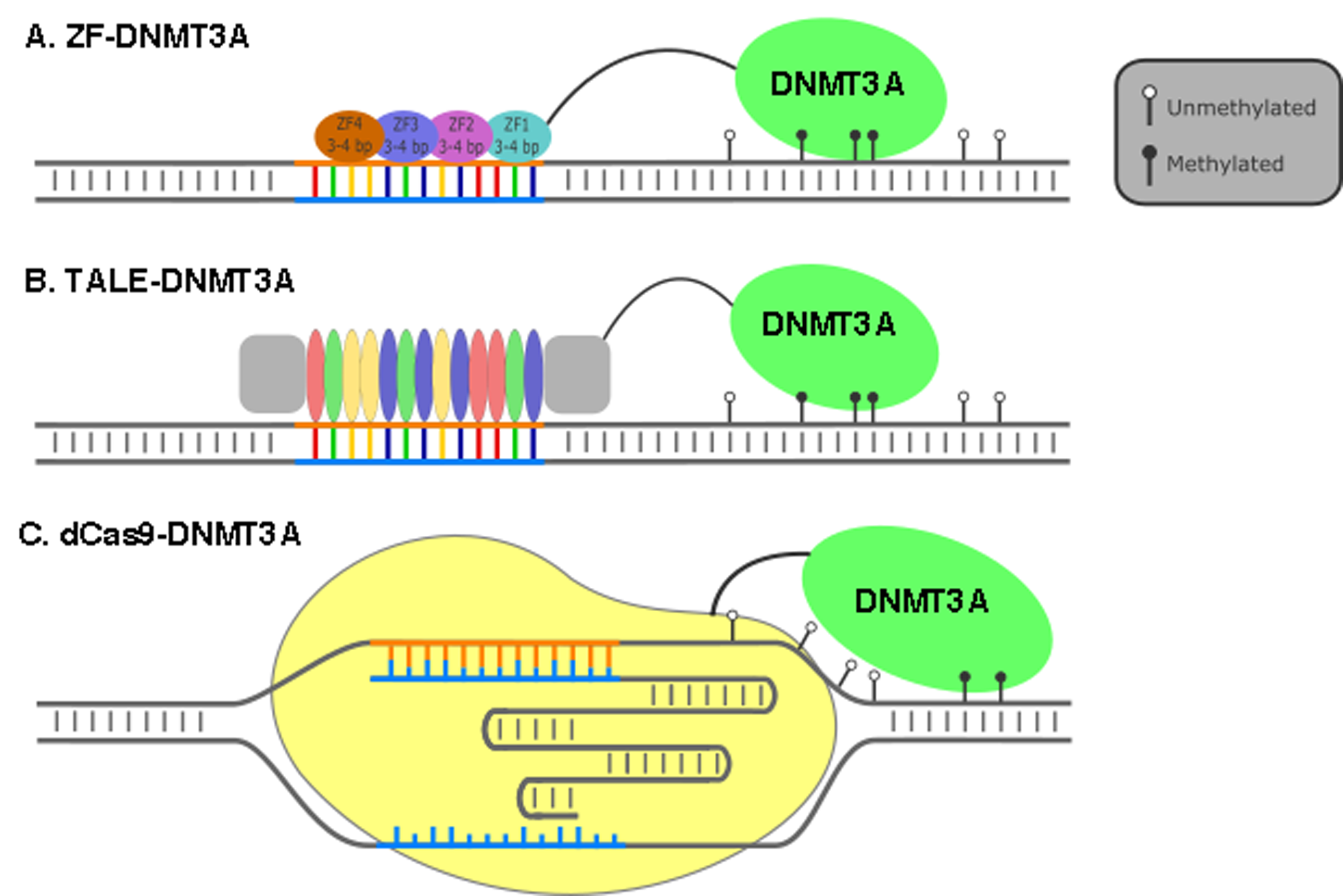 Figure 1. Schematic Illustration of Programmable Epigenetic Editing tools for targeted DNA methylation.
Figure 1. Schematic Illustration of Programmable Epigenetic Editing tools for targeted DNA methylation.